



研究開発段階から育て上げる「sawai品質」
製剤は出荷する前に必ず出荷試験が実施されますが、 当社では出荷試験による品質の保証に加え、研究開発段階から「sawai品質」を作り込んでいくことを大切に考えています。今、当社では、新しいジェネリック医薬品の研究開発に着手すると同時に「sawai品質」の設計に取り掛かり、研究開発を通して育て上げ、製品に作り込んでいます。その過程には、関連する各種ガイドラインで求められる品質に加え、科学と技術を品質に練り込んでいく研究員のこだわりとカルチャーが詰まっています。
医薬品原薬の選定
当社が、医薬品づくりにおいて非常に重視していることの1つが、原薬の選定です。沢井製薬では世界中から原薬を求め、基本的に厚生労働省が定める規格より厳しい自主基準に適合した原薬を選択すると同時に、高い製剤品質を守るため、原薬の段階からその物性(性質や状態)などについても徹底的に分析しています。
不純物の管理
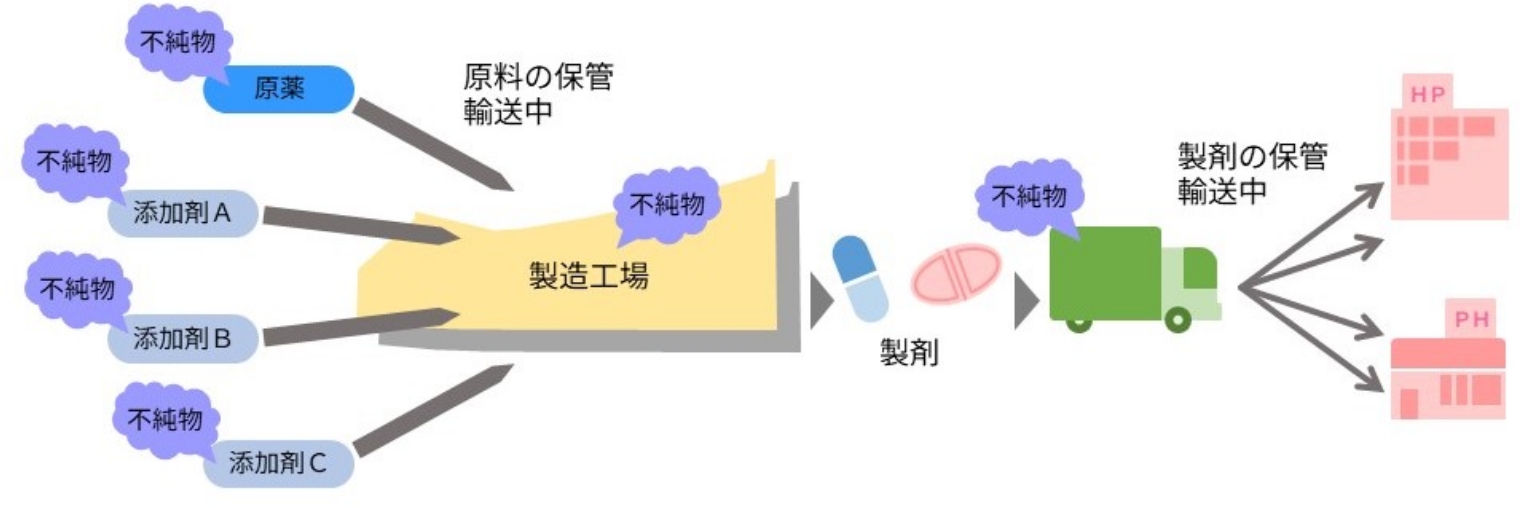
製剤の原料(原薬及び添加剤)には、原料自体や原料の生成に使用される試薬の品質、その製造工程(化学反応、抽出・精製、細胞培養・発酵、酵素反応など)に由来する様々な不純物が混入するリスクがあります。当社は、安全性の観点から、混入する可能性のある不純物を同定し、その量を様々なガイドラインに従って厳格かつ適正に管理しています。また、製造工程及び原料や製剤の保管中に原料が分解して不純物が生成されるリスクもあるため、保管に関しても同様です。
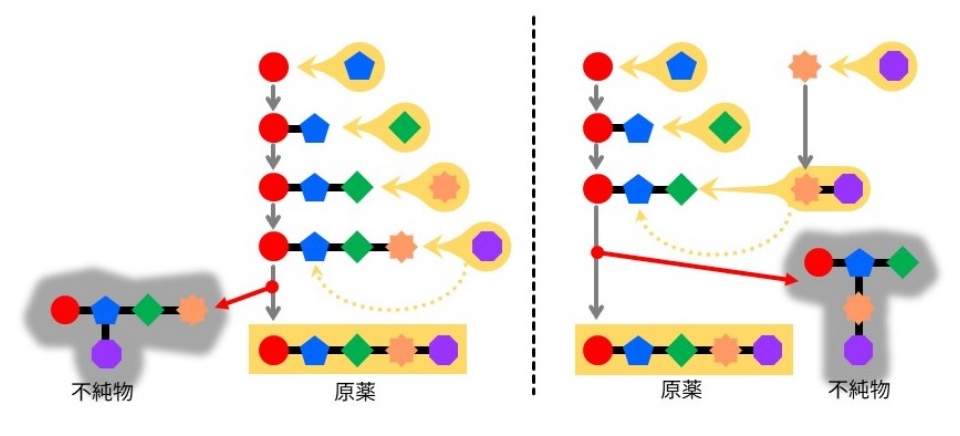
■合成ルートの確認
原薬は原薬メーカーごとに製造方法が異なる場合が多く、その影響で原薬に混入する可能性のある不純物も異なってきます。当社では、原薬に実際に混入している不純物のみならず、合成ルートから考えられる潜在的な不純物を予測し、原薬に混入する可能性のリスク評価を行っています。また、ICH※1のガイドライン※2に示される、原薬の出発物質選定基準と照らして出発物質の妥当性評価を行い、上流工程からの品質リスク評価を踏まえて原薬メーカーと出発物質を再設定する必要性を協議します。
※1 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use(医薬品規制調和国際会議)
※2 原薬の開発と製造(化学薬品及びバイオテクノロジー応用医薬品/生物起源由来医薬品)ガイドライン ICH Q11
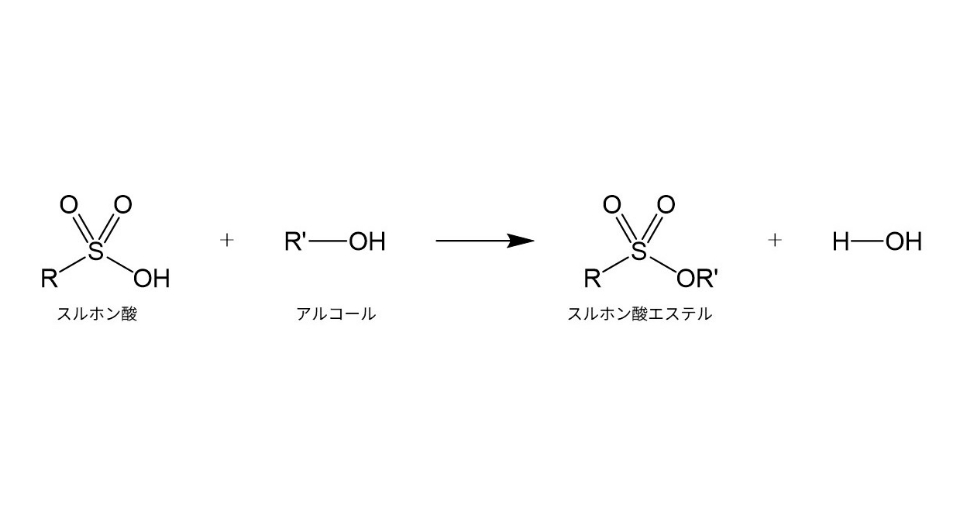
■残留溶媒
化学合成で製造される原薬は、製造工程において有機溶媒を使用するため、原薬に溶媒が残留するリスクがあります。残留溶媒はICHのガイドライン※3で溶媒ごとの残留許容値が設定されています。原薬メーカーは、製造方法からガイドライン※3に従って残留溶媒の規格を設定し、その情報が品質試験成績書に記載されていますが、当社でも評価を行い、品質試験成績書に記載されていない残留溶媒の有無についても厳しくチェックしています。
また、残留溶媒に起因して毒性が強い不純物が生成する場合があります。例えば、原薬がスルホン酸類との塩である場合、アルコール系溶媒と反応して変異原性を有するスルホン酸エステル類を生成する可能性があるため、アルコール系溶媒が残留する際には原薬製造中や原薬、製剤の保存中について特に注意を払います。
※3 医薬品の残留溶媒ガイドライン ICH Q3C
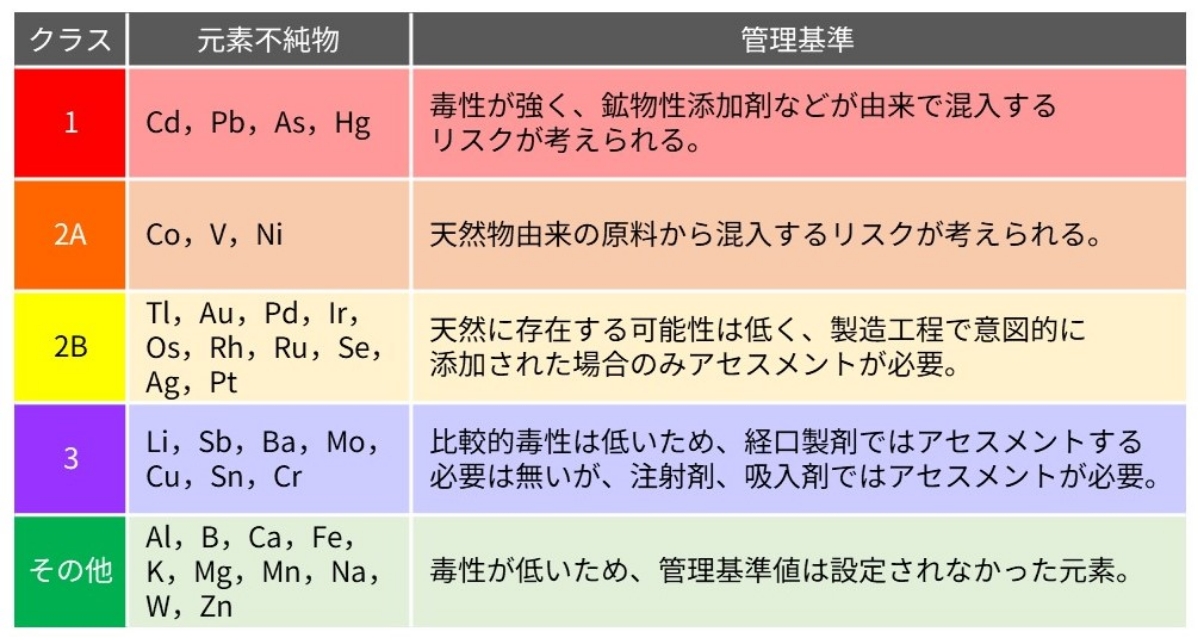
■元素不純物
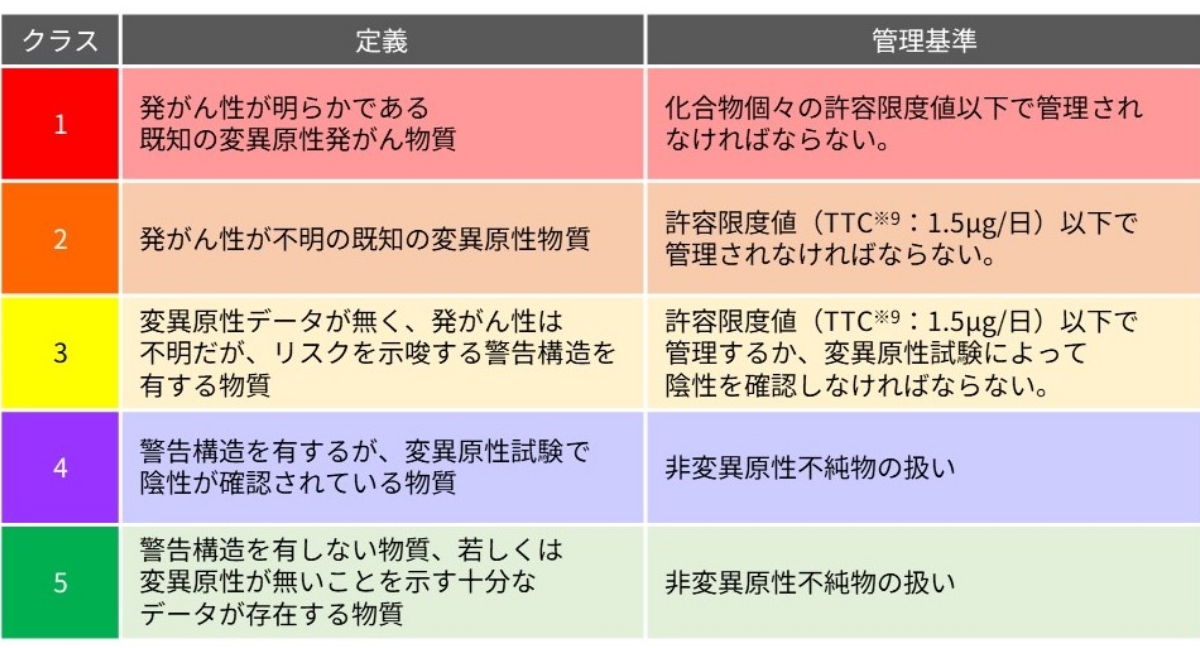
生体に作用する微量元素で、特に有害性を持つ金属元素を主に指し、ICHのガイドライン※4では医薬品中の元素不純物の管理について規定されています。ガイドライン※4では、各元素の毒性、投与経路に基づいた限度値が設定され、毒性と混入する可能性に基づいてクラス分けがされています(図1)。
※4 医薬品の元素不純物ガイドライン ICH Q3D
元素不純物は、原薬や製剤の保存中に新たに生成したり、増加したりするものではないため、製造工程で使用される原料、資材、設備から混入するリスクをアセスメントする必要があります。
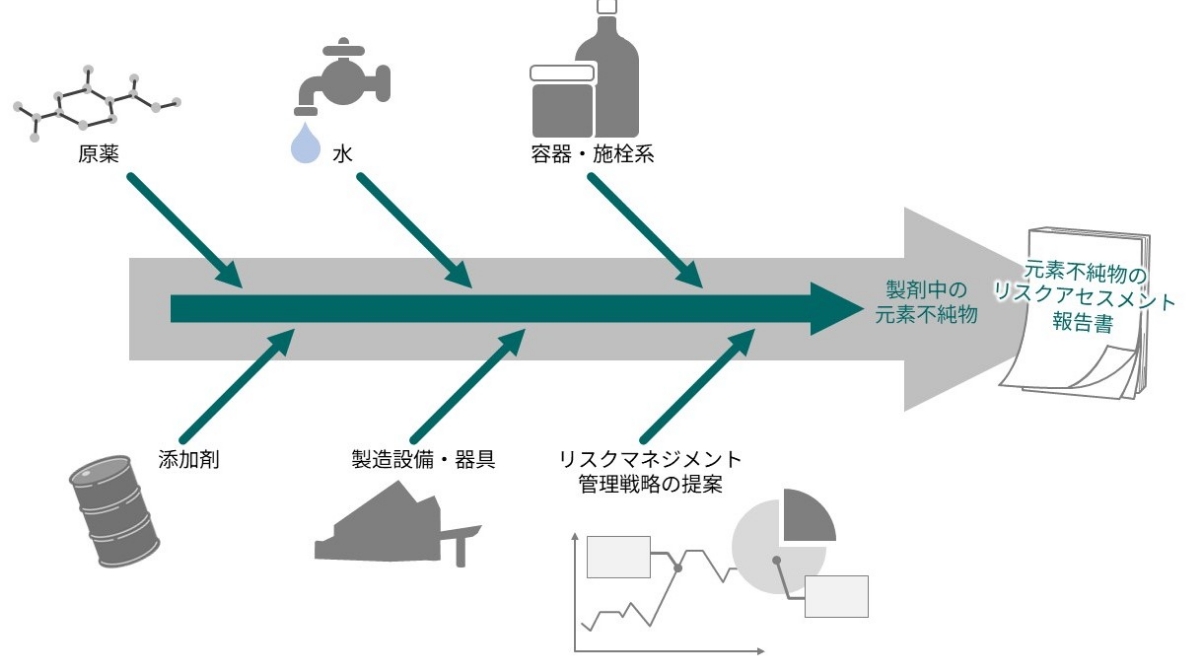
当社では、エネルギー分散型蛍光X線分析装置(EDX)や誘導結合プラズマ質量分析装置(ICP-MS)といった分析装置を用いて、原薬や製剤中の元素不純物を測定しています。EDXでは主に混入元素の有無の確認に用い、ICP-MSでは微量に含まれる元素を高感度(EDXの1,000,000倍)で測定し、多面的な評価を実施しています(図2)。

■類縁物質
高速液体クロマトグラフィー(HPLC)やガスクロマトグラフィー(GC)などの分析機器を用いて、原薬に含まれる類縁物質を調査します。原薬に含まれる類縁物質を見逃さないために、より多くの類縁物質を広範囲で確認できる2つの試験条件を確立します。得られた類縁物質の情報をICHのガイドライン※5や先発品の類縁物質プロファイル※6に照らして評価し、基本的に先発品と同等以上の品質基準を設定して管理しています。未知の類縁物質が検出された場合には、液体クロマトグラフィー質量分析計(LC-MS)やガスクロマトグラフィー質量分析計(GC-MS)などの精密な分析機器を用いて類縁物質の質量を特定し、製造方法から考えられる構造式を推定します。必要に応じて類縁物質を分取して、 核磁気共鳴装置(NMR)などの分析機器を用いて解析し、構造式を決定します。決定した構造式からその類縁物質の発生源を調査し、許容値以下に制御するために原薬メーカーに管理戦略を確認しています。
特にペプチド医薬品の開発においては、類縁物質が生物活性又は免疫原性、オフターゲット効果※7を示す可能性が考えられるため、最新の分析機器を用いて類縁物質を特定し、生化学的評価を行い、自社製剤の生物活性又は免疫原性、オフターゲット効果※7のリスクが先発品と同等/同質であることを確認しています。
※5 新有効成分含有医薬品のうち原薬の不純物に関するガイドライン ICH Q3A
※6 原薬中に存在する構造既知又は未知の不純物の全体像
※7 本来の標的(オンターゲット)とは異なる分子(オフターゲット)を阻害あるいは活性化してしまう効果
■変異原性不純物
変異原性不純物は、ヒトの遺伝情報に変化を引き起こす作用(変異原性)を有し、ヒトにがんを引き起こす不純物であり、ICHのガイドライン※8で厳格な管理が要求されています(図3)。
※8 潜在的発がんリスクを低減するための医薬品中DNA反応性(変異原性)不純物の評価及び管理 ICH M7
※9 TTC(threshold of toxicological concern:10-5という理論上の生涯過剰発がんリスクに相当する毒性学的懸念の閾値以下)
当社では、外部専門家と共に全開発品目の変異原性不純物のリスクアセスメントを実施しています。リスクアセスメントは以下の手順1)~4)に従って実施します。
1)構造活性相関による変異原性予測ソフトを用いた評価(QSAR評価)
ICH M7ガイドラインに従って知識ルールベースと統計ベースからなる2つの変異原性予測ソフトを用いて評価します。評価対象は、原薬の製造に使用される原料、中間体、試薬、溶媒、さらに合成ルートから予測される不純物まで広範囲に評価します。
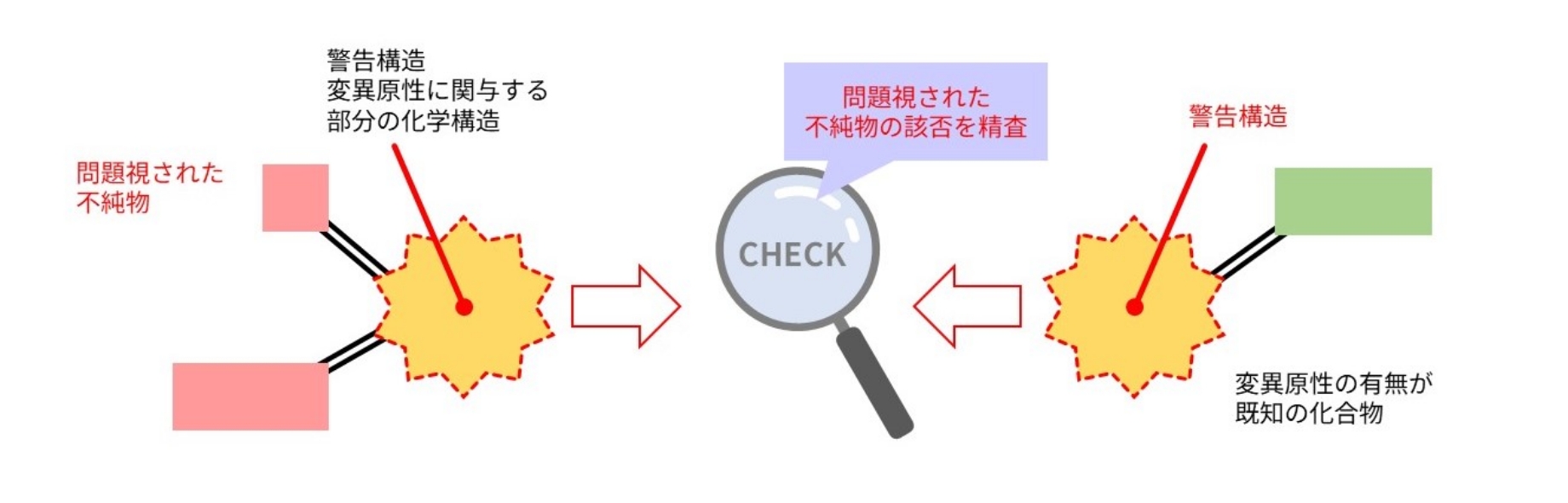
2)QSAR評価で得られた変異原性警告構造について精査
1)で問題視された不純物について、すでに変異原性の有無が確認されている類似する化合物から、当該不純物の該否を精査します。
※横スクロール出来ます。
3)原薬メーカーから入手したリスクアセスメントレポートを確認
4)外部専門家を交えて変異原性不純物の管理戦略の妥当性を確認
自社でのQSAR評価1)、2)を踏まえ、原薬メーカーのリスクアセスメントや管理戦略の妥当性について外部専門家を交えて確認します。
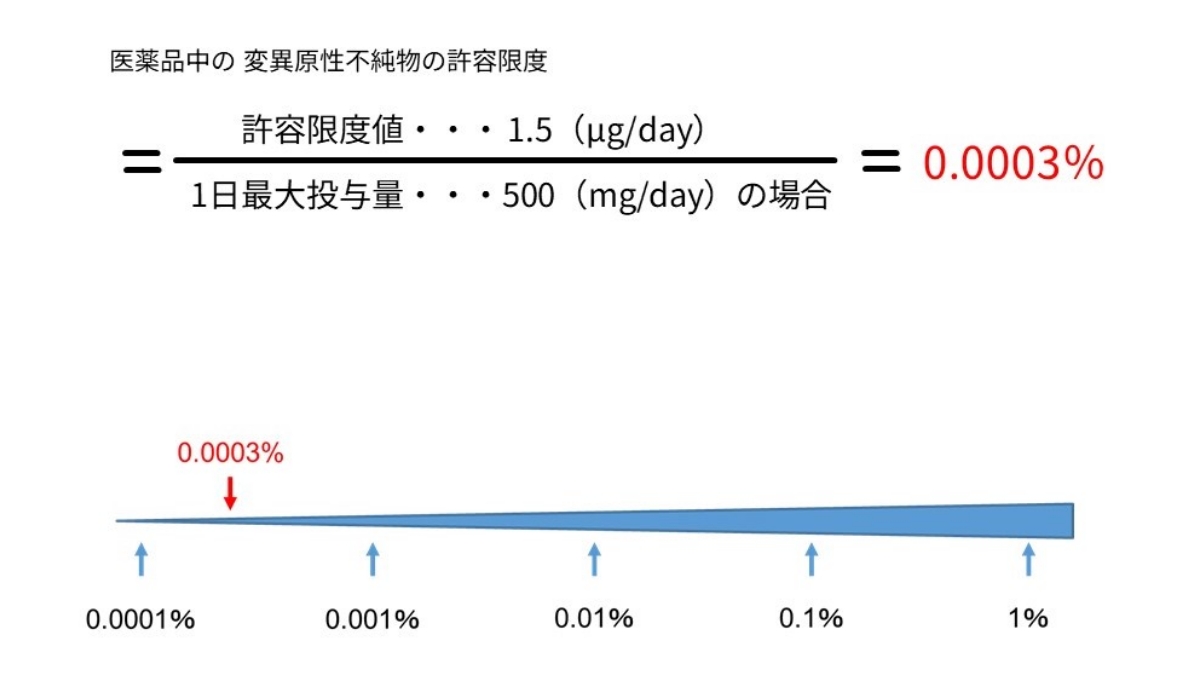
ここで、実際の医薬品中の変異原性不純物の管理法について触れたいと思います。例えば、1日最大投与量が500mgの医薬品中の変異原性不純物を許容限度値(TTC※9) 1.5µg/日以下で管理する場合、以下の計算式により換算され、微量の管理(0.0003%以下)が必要となります。
当社では、微量の変異原性不純物を管理するために、0.0000001%レベルまで測定可能なLC-MSを用いて高感度の試験法を確立し、変異原性不純物を厳格に管理しています。
ニトロソアミン類
TTC ※9(1.5µg/day)を下回る摂取量であっても理論的に著しい発がんリスクの可能性を伴うことが確認されている変異原性不純物は、ガイドライン※7で「cohort of concern」に分類され、不純物個々に厳格に管理されます。
中でもニトロソアミン類は、過去に医薬品から検出された事例もあり、各製薬企業に自主点検が求められています。
当社では「反応性NOx」という新しい管理指標概念を考案し、ニトロソアミン類の生成を抑制した製品開発に挑戦しています。
製剤特性に影響を及ぼす原薬物性の評価
不純物の管理だけでなく原薬の結晶形や粒子径といった様々な物性についても製剤化に適したものであることを慎重に検討する必要があります。
原薬の多くは結晶性であり、複数の結晶形(結晶多形)を示すことがあります。結晶多形を粉末X線回折(図4)、固体NMR(図5)、熱分析(図6)などの分析機器を用いて解析し、製剤化に適した結晶形を選択しています。選択した結晶形が製剤化工程や製造後の保存過程で別の結晶形に転移(結晶転移)するリスクがある場合には、使用期限内に結晶転移を起こさないように製剤設計しています。
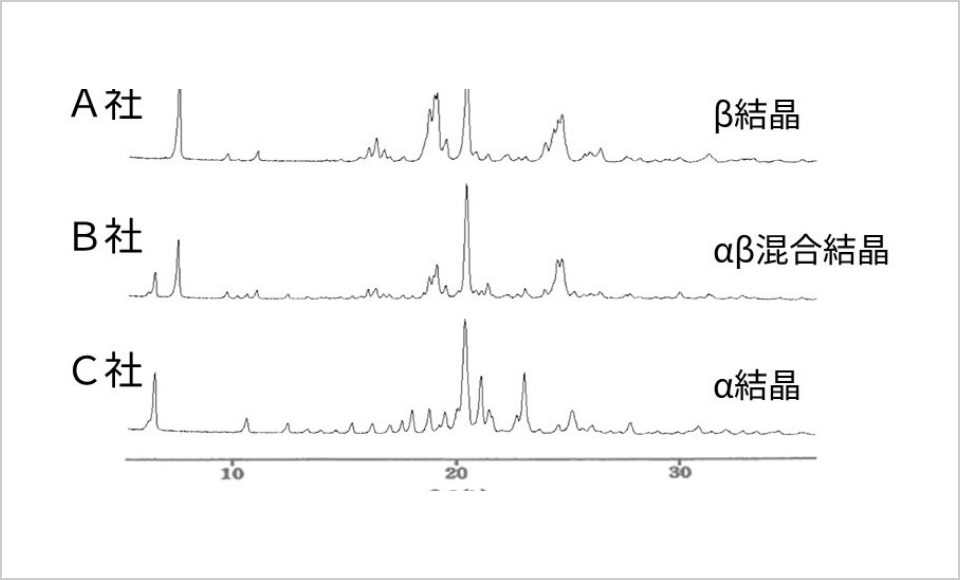
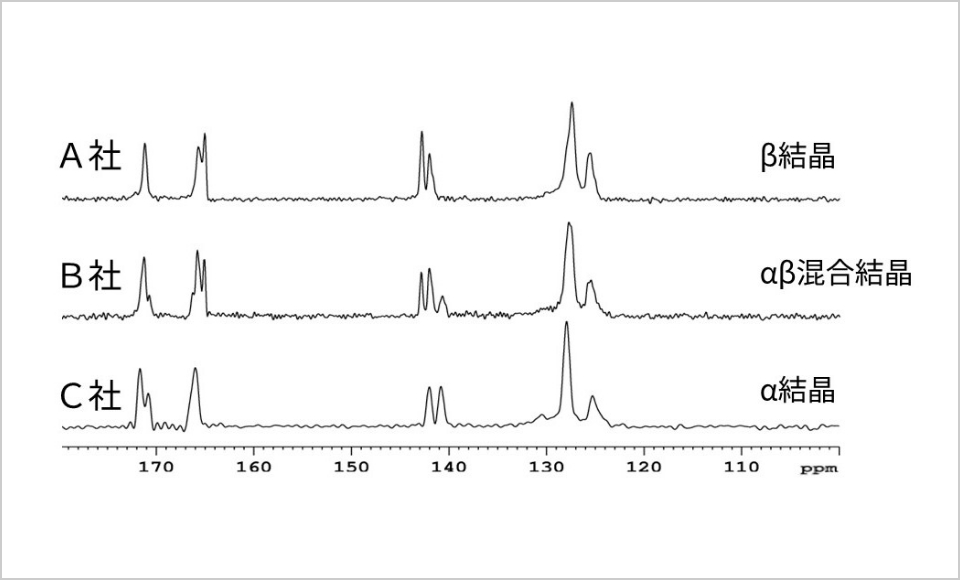
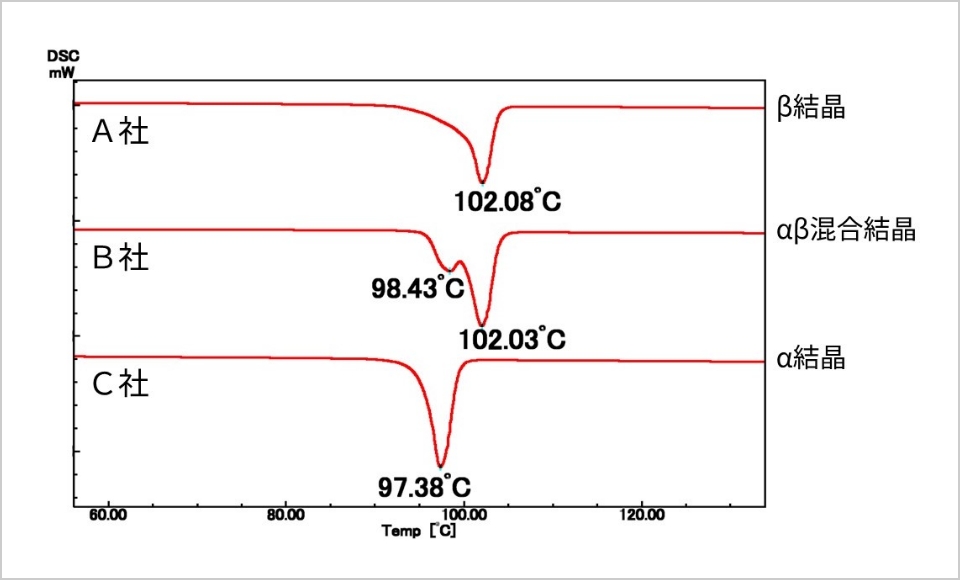
また、原薬の粒子径は、製剤化工程の製造性、製造後の溶出性や安定性に影響することがあるため、適切な粒度分布幅を設定して管理しています。粒度試験法は分析装置の原理や機種、試験方法の違いにより、測定値が異なる場合があるため、同一ロットで測定値が合うように試験方法を検討した上で粒子径評価を行います。
結晶形や粒子径以外にも比表面積やかさ密度、ぬれ性の違いなど様々な物性についても走査電子顕微鏡(SEM)、画像式粒度分布測定装置やパルスNMRを用いて多面的に評価します。このような評価は複数の原薬メーカーで製造販売承認を取得する場合も重要になることがあります。
このように、当社は研究開発着手時点から様々な角度で検討を行い、sawai品質を作り込んでいます。
(2024年3月作成)